

Research
The Lilly and Blair Foundation is not just funding research; we are architecting a cure. To move from the laboratory to a human clinical trial, we drive a coordinated attack across three strategic pillars:
Therapeutic Development: We are engineering the "medicine" itself, utilizing gene editing, ASOs, and drug repurposing to fix the genetic root of SPG4.
Modeling and Testing Platforms: We build the living laboratories—from humanized mice to 3D organoids—required to prove these medicines are safe and effective.
Clinical Trial Readiness: We are building the regulatory bridge, establishing the blood tests and natural history data the FDA requires to approve a cure.
Therapeutic Development
Modeling and Testing Platforms
-
![]()
The Humanized R499H Mouse Model
The world’s first living roadmap for the most severe SPG4 mutation.
Background: To test modern genetic medicines like ASOs or Gene Editors, the laboratory model must carry the exact human genetic sequence. Standard mice do not have the human SPAST gene, making them "invisible" to these precision drugs.Strategy: We invested in the creation of a "humanized" model—a mouse engineered to carry the human SPAST gene with the severe R499H mutation found in many children.
Solution: Fully funded by the Lilly and Blair Foundation, this model is now the global "gold standard" for testing. It allows us to validate "clinic-ready" therapies in a living system that mimics human biology.
Impact: This model removes the guesswork from drug development, providing the high-level safety and effectiveness data required to gain FDA clearance for human trials.
-
![]()
Large Animal Science: SPG4 Cattle
Bridging the gap from lab mice to human reality.
Background: A mouse’s spinal cord is only a few centimeters long. A human child’s nervous system is vastly larger and more complex. Dosing and delivery methods that work in a tiny mouse often fail to scale up to a human.
Strategy: We maintain a dedicated herd of SPG4-affected cattle at Hillcrest Farm. Because their size and spinal cord complexity are closer to a human’s, they serve as a vital "natural model."
Solution: By studying how therapies travel through the nervous system of these large animals, we can refine our delivery techniques (such as IV or spinal injections) to ensure the medicine reaches the target nerves in a human child.
Impact: The insights gained from Hillcrest Farm ensure that we have the right "map" and the right dosage before we move into the clinic, significantly increasing the chances of trial success.
-
![]()
3D Brain Organoids
Creating a living laboratory from a patient’s own cells.
Background: To find the right treatment, we need to see how human neurons behave. Standard cell cultures don't capture the complex "wiring" of the human brain.
Strategy: The Qiang Lab (Drexel), has grown 3D Brain Organoids (mini-brains) from the stem cells of children with the R499H mutation.
Solution: These organoids have the exact DNA of some of our children, serving as the "engine" for drug repurposing screens and providing a human environment to test gene therapies.
Impact: This platform allows us to see how human nerves react to treatments in real-time, providing proof-of-concept for every therapy in our pipeline.



Clinical Trial Readiness
-
![]()
The NULISA Study: Biomarker Discovery
Establishing the blood test that will unlock FDA approval.
Background: The FDA will not approve a drug unless we can prove it is working through "biomarkers"—measurable indicators in the blood. Historically, no such marker has existed for SPG4.Strategy: Fully funded by The Lilly and Blair Foundation, Boston Children’s Hospital is utilizing the ultra-sensitive NULISAseq platform to scan for a unique "protein fingerprint" in the blood of SPG4 patients.
Solution: BCH is analyzing longitudinal samples to identify specific proteins that change as the disease progresses. This will create a scalable blood test to monitor treatment response.
Impact: A validated biomarker allows for shorter, more efficient clinical trials. Instead of waiting years to see if a child's walking improves, we can see if the therapy is working in weeks via a simple blood test.
-
![]()
The Natural History Studies: SP-CERN and Pediatric Tracking
Building the definitive roadmap for SPG4 across a lifetime.
Background: To cure a disease, you must first define it. Without a clear understanding of how SPG4 progresses from the first sign of symptoms through adulthood, regulators like the FDA cannot determine if a new treatment is truly effective.
Strategy: We support the global registry and longitudinal study led by Dr. Darius Ebrahimi-Fakhari at Boston Children’s Hospital (NCT04712812). This network connects researchers worldwide to track the lived experience of SPG4 patients of all ages.
Solution: SP-CERN collects standardized clinical assessments, advanced gait analysis, and bio-samples over many years. This creates a "Master Database" that defines the disease's trajectory and identifies the specific windows of time where treatments will be most impactful.
Impact: This work transforms SPG4 from a "rare unknown" into a "trial-ready" disease. It provides the essential "baseline" data that scientists, clinicians, and regulatory bodies need to design, validate, and approve the clinical trials of tomorrow.
Learn more and enroll » -
![]()
The SPG4 Biobank: Coriell Institute
Providing the biological "building blocks" for global research.
Background: Researchers cannot study SPG4 without access to patient cells. A centralized biobank ensures that scientists everywhere have the samples they need to innovate.
Strategy: We encourage families to contribute blood or skin samples to the NIGMS Repository at the Coriell Institute, an international biobank.
Solution: These samples are the foundation of our work; for example, the 3D Brain Organoids grown at Drexel were created using samples from this very biobank.
Impact: By donating a sample, families provide the raw materials for a thousand different experiments, ensuring that SPG4 research never hits a "supply chain" bottleneck.
-
![]()
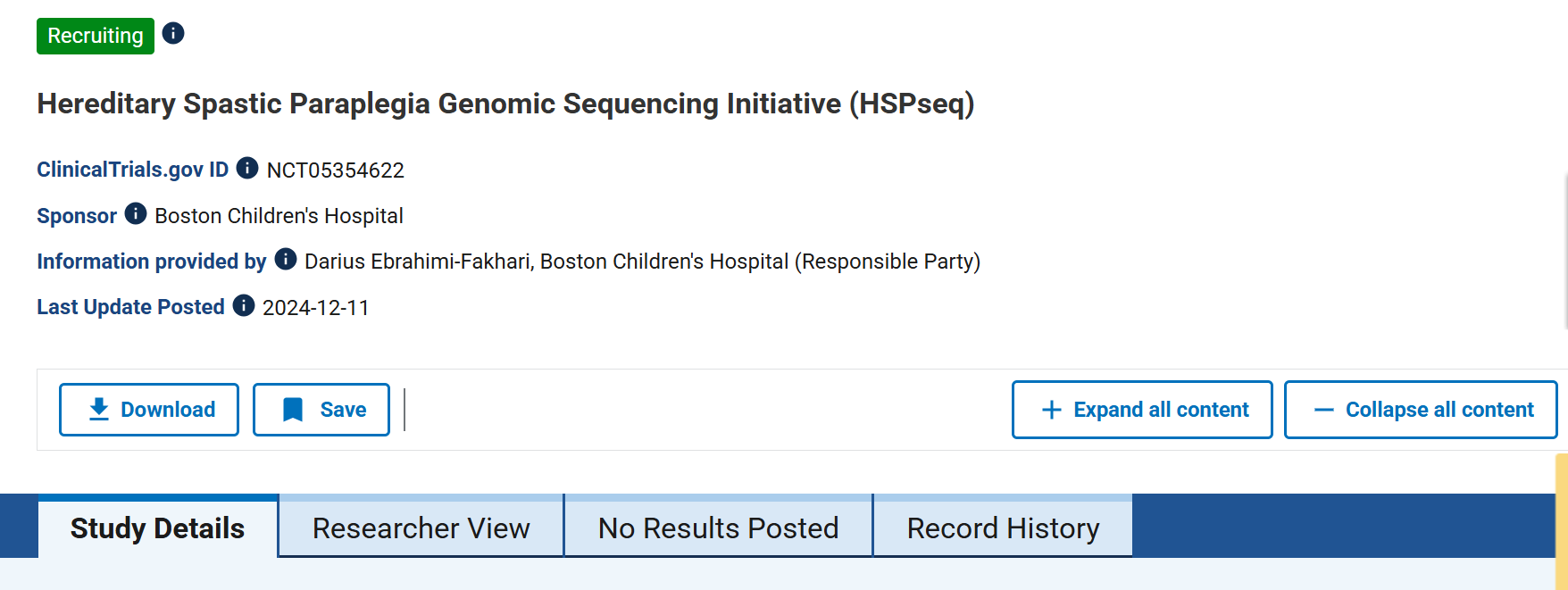
HSPseq: Genomic Sequencing Initiative
Unlocking the full genetic spectrum of childhood spasticity.
Background: Not every child with spasticity has a clear answer. Defining the full genomic landscape of HSP is essential for personalized medicine and early diagnosis.
Strategy: Led by Boston Children’s Hospital, the HSPseq study combines clinical data with advanced genomic sequencing for patients aged 1 month to 30 years.
Solution: This initiative identifies new genes and better defines the spectrum of SPG4, helping families get answers sooner—regardless of whether they have a known or unknown genetic diagnosis.
Impact: Earlier diagnosis means earlier intervention. HSPseq ensures that as new therapies come online, every child is correctly identified and ready to receive treatment.
Learn more and enroll »



Research Collaborations
-

Darius Ebrahimi-Fakhari, MD, PhD
-

Peter Baas, PhD
Liang Oscar Qiang, MD, PhD
Emanuela Piermarini, PhD -

Miguel Sena-Esteves, PhD
Heather Gray-Edwards, DVM, PhD -

Anjon Audhya, PhD
Molly Lettman, PhD
Foundation and Industry Partners






What happens next depends on how quickly we move.
Your support accelerates progress toward real treatments for children with SPG4.